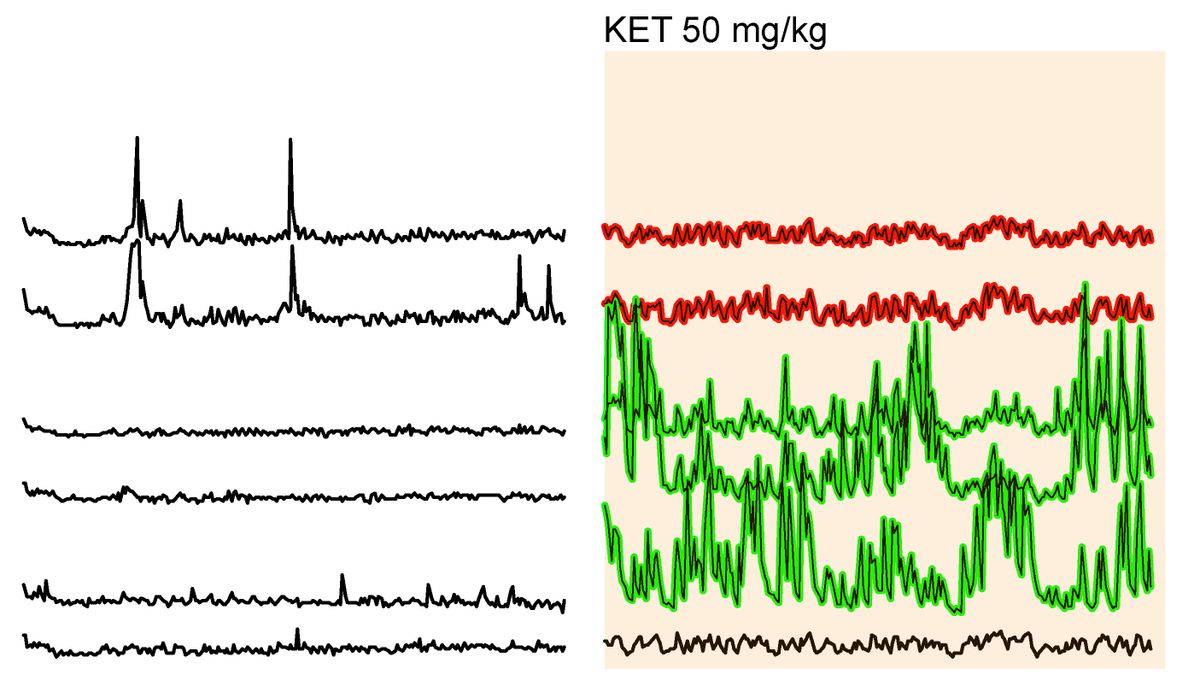
Thanks to continued weekly medications, a 16-month-old girl shows no symptoms of a severe genetic disease that typically kills children before they turn two.
In a medical first, an infant was successfully treated for a rare, fatal genetic disease before she was born, via therapeutic enzymes injected into her umbilical cord.
The girl, now 16 months old and symptom-free, according to a case study published yesterday (November 9) in The New England Journal of Medicine, has a severe form of a rare genetic condition called infantile-onset Pompe disease. People with this disease produce too little or no acid alpha-glucosidase (GAA), an enzyme that allows lysosomes to break glycogen down into usable glucose. Without GAA, glycogen deposits build up, causing irreversible, progressive damage to the heart and other muscles.
If the condition is left untreated, children with the most severe form of the disease (which affects fewer than one in 100,000 babies, according to STAT News) accumulate this damage before they’re even born, and rarely live to see their second birthdays.
The only existing treatment for Pompe disease, regardless of severity, is enzyme replacement therapy (ERT) to supply the missing GAA, together with an immunosuppressant to prevent rejection by the immune system. But this has so far only been used after birth. In some milder cases, diagnosis may not happen until years or decades later, according to The New York Times, delaying treatment. Thanks to the prenatal therapy, Ayla has developed normally, met the developmental milestones expected of a child not afflicted by Pompe, and exhibited healthy biomarkers, growth, and behavior.
Before the advent of ERT, which for years has staved off further heart damage in infants diagnosed after birth, “we used to see babies with infantile Pompe, . . . The only information we could give the parents was, ‘This is a lethal disease. Go home and enjoy your baby,’” Duke University researcher Priya Kishnani, who originally developed ERT for Pompe disease and is a coauthor of the case study, tells the Times.
In this case, Ayla’s parents had already lost two children to severe Pompe disease, so they sought out prenatal testing, according to STAT. The Times reports that the parents were referred to Tippi MacKenzie, a pediatric surgeon at the University of California, San Francisco, who contributed to the NEJM case study. MacKenzie had authorization to conduct a clinical trial on prenatal ERT, so she arranged for the family to receive treatment in Ottawa, where they live, as the COVID-19 pandemic prevented them from traveling. Doctors there performed six infusions of the therapeutic enzyme directly into the growing fetus’s umbilical cord between gestation weeks 24 and 36.
See “How COVID-19 Affects Pregnancy”
The doctors found no indication of the dangerous heart thickening that had claimed the lives of the girl’s two siblings. “It’s a great step forward,” Children’s Hospital of Philadelphia pediatric and fetal surgeon Bill Peranteau, who wasn’t involved in the work, tells Science News.
Ayla continues to receive ERT every week, as the prenatal treatment prevented cardiomyopathy but cannot cure the disease. “It’s not like we’re saying, ‘Hey, this is a checked box and a success story,’” Kishnani tells STAT. “It is looking very promising, but I still think we’re going to follow this child very carefully.”
Still, the study authors say their success indicates that prenatal ERT could improve postnatal outcomes in other diseases, according to the Times, and the same group behind the NEJM case study says it has already treated a fetus diagnosed with Hunter syndrome in a similar fashion.